
Josh Milthorpe
Computer Scientist, Oak Ridge National Laboratory
Resolutions of the Coulomb operator: VIII. Parallel implementation using the modern programming language X10
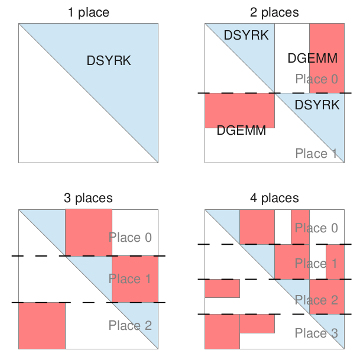
Use of the modern parallel programming language X10 for computing long-range Coulomb and exchange interactions is presented. By using X10, a partitioned global address space language with support for task parallelism and the explicit representation of data locality, the resolution of the Ewald operator can be parallelized in a straightforward manner including use of both intranode and internode parallelism. We evaluate four different schemes for dynamic load balancing of integral calculation using X10’s work stealing runtime, and report performance results for long-range HF energy calculation of large molecule/high quality basis running on up to 1024 cores of a high performance cluster machine.
GitHub repository: ANUChem (see application: Pumja Rasaayani)